
Without a doubt, introducing immune checkpoint inhibitors to the clinic was a major breakthrough in the war against cancer. For some patients tumor responses to anti-PD-1/PD-L1 or anti-CTLA4 therapies are spectacular and last long after the therapy is withdrawn. Interestingly, disease regression can occur even after an initial phase of tumor growth during the therapy. However, despite spectacular successes, therapies based on checkpoint inhibitors still suffer from relatively low response rates. There is an urgent need to establish reliable patient-specific biomarkers that would allow to solve this problem. The current focus is on using, among others, measures of pretreatment T cell infiltration, PD-L1 expression profiles, inflammatory status, and mutational burden of the tumor. Finding a reliable correlate will allow for designing patient-specific therapy scheduling or for incorporating appropriate adjuvant treatment.
In todays post, I will adopt a mathematical model that was introduced by Kuznetsov and colleagues back in 1994 (Kuznetsov et al., Bull Math Biol, 1994) to investigate the effects of anti-PD-1/PD-L1 therapy depending on the pretreatment tumor characteristics. The way in which the model was formulated more than two decades ago allows for elegant and straightforward incorporation of such a treatment. I will show how the variability in the observed response dynamics is explained by the model and how can we estimate the minimal time on PD-1/PD-L1 treatment required for a therapeutical success.
1. Mathematical model and its predictions
In his paper Kuznetsov and colleagues introduced the following relatively simple mathematical model describing tumor-immune system dynamics
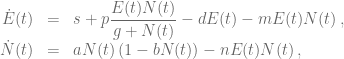
where 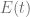

First, we should check what is the predicted model dynamics for the nominal parameter values proposed by Kuznetsov and others. Let us simplify that task by performing non-dimensionalization procedure first. After defining 
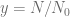

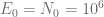
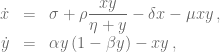
where 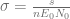
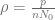
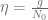
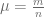
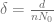
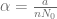

Using the parameters proposed in Kuznetsov’s original paper we obtain the following values of non-dimensional parameters:

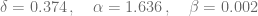
We can start our analysis with evaluating numerically the model behavior for the above set of parameters. In order to do that systemically we will plot the phase portrait in which behavior of the solutions in the whole phase space can be viewed. MATLAB is quite a convenient environment in which we can do that quite easily by using the built-in quiver function:
%% DEFINING MODEL PARAMETERS
sigma = 0.118; rho = 1.131; eta = 20.19; mu = 0.00311;
delta = 0.374; alpha = 1.636; beta = 0.002;
%% DEFINING THE MODEL (INLINE FUNCTION)
rhs = @(t,x)([sigma+rho*x(1,:).*x(2,:)./(eta+x(2,:))-mu*x(1,:).*x(2,:)-delta*x(1,:);...
alpha*x(2,:).*(1-beta*x(2,:))-x(1,:).*x(2,:)]);
%% FUNCTION RETURNING MODEL SOLUTION ON [0,100] FOR GIVEN INITIAL CONDITION
solve = @(init)(ode45(rhs,[0 100],init));
%% EVALUATING THE VECTOR FIELD IN [0, 3.5]x[0, 450]
Npoints = 30;
x = linspace(0.,3.5,Npoints);
y = linspace(0,450,Npoints);
dx = x(2)-x(1);
dy = y(2)-y(1);
[X, Y] = meshgrid(x,y);
G = rhs([],[reshape(X,1,[]); reshape(Y,1,[])]);
U = reshape(G(1,:),Npoints,Npoints);
V = reshape(G(2,:),Npoints,Npoints)*dx/dy;
N = sqrt(U.^2+V.^2);
U = U./N; V = V./N;
%% EVALUATING MODEL SOLUTIONS FOR DIFFERENT INITIAL CONDITIONS
initCond = [0, 2.58; 0, 1; 0, 0.1; 0, 20; 0.76, 267.8;...
0.755, 267.8; 1.055, 450; 0.6, 450; 2.1, 450];
sols = cell(1,size(initCond,1));
for i = 1:size(initCond,1)
sols{i} = solve(initCond(i,:));
end
%% PLOTTING THE RESULTS
figure(1)
clf
hold on
[X1, Y1] = meshgrid(0:Npoints-1,0:Npoints-1);
q = quiver(X1,Y1,U,V); %plotting vector field
q.Color = 'red';
q.AutoScaleFactor = 0.75;
for i = 1:length(sols) %plotting each solution
plot(sols{i}.y(1,:)/max(x)*(Npoints-1),sols{i}.y(2,:)/max(y)*(Npoints-1),'k')
end
hold off
After running the above script we should see an image similar to the one below.
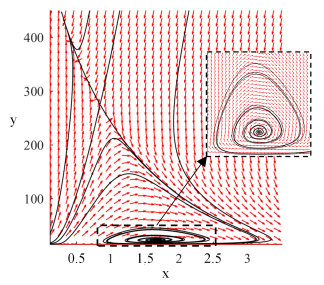
We can see in the phase portrait that there are two steady states to which solutions tend to, one with substantial tumor burden, i.e. with large value of variable y, the second reflecting probably subclinical disease. Of course, those are the initial conditions that govern to which of them the solution will tend to. Let us now check analytically if the above behavior is not a numerical artifact.
First of all, the above system has a smooth vector field and thus, solutions exist and are unique. Moreover, it is easy to check that the solutions are bounded and non-negative. We are interested in calculating all of the possible steady states of the system and evaluating their stability. For any set of parameters we always have a semi-trivial steady state 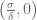
where

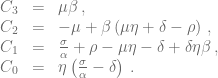
We can see that, as we have cubic polynomial, there are up to three additional steady states. The exact number of solutions depends obviously on particular 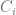
The local stability of the steady states can be evaluated by checking the eigenvalues of Jacobian matrix for the considered system:
![J(x,y) = \left[ \begin{array}{cc} \frac{\rho y}{\eta + y}-\mu y-\delta & -\mu x + \frac{\rho \eta x}{(\eta + y)^2} \\ -y & \alpha\left(1-2\beta y\right)-x \\ \end{array} \right].](https://s0.wp.com/latex.php?latex=J%28x%2Cy%29+%3D+%5Cleft%5B+%5Cbegin%7Barray%7D%7Bcc%7D+%5Cfrac%7B%5Crho+y%7D%7B%5Ceta+%2B+y%7D-%5Cmu+y-%5Cdelta+%26+-%5Cmu+x+%2B+%5Cfrac%7B%5Crho+%5Ceta+x%7D%7B%28%5Ceta+%2B+y%29%5E2%7D+%5C%5C+-y+%26+%5Calpha%5Cleft%281-2%5Cbeta+y%5Cright%29-x+%5C%5C+%5Cend%7Barray%7D+%5Cright%5D.+&bg=ffffff&fg=5e5e5e&s=0&c=20201002)
If all eigenvalues of 
An additional step that will be further required to draw definite conclusions is to show that there are no cycles in the system, i.e. all solutions tend to one of the steady states. We can do that by using Dulac-Bendixon theorem with the function defined as 
We are interested in behavior of the system for different values of parameter 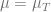
%% DEFINING MODEL PARAMETERS sigma = 0.118; rho = 1.131; eta = 20.19; mu = 0.00311; delta = 0.374; alpha = 1.636; beta = 0.002; %% DEFINING FUNCTION RETURNING JACOBIAN MATRIX FOR GIVEN x,y AND mu J = @(x,y,mu)([rho*y/(eta+y)-mu*y-delta, -mu*x+rho*eta*x/(eta+y)^2 ; ... -y, alpha*(1-2*beta*y)-x]); %% DEFINING POLYNOMIAL COEFFICIENTS FOR DIFFERENT MU C = @(mu)( [mu*beta; -mu+beta*(eta*mu+delta-rho); ... sigma/alpha+rho-eta*mu-delta+beta*delta*eta; ... eta*(sigma/alpha-delta)]); %% PLOTTING BIFFURCATION DIAGRAM muMesh = linspace(0,5*mu,100); %mesh for mu figure(1) clf hold on for mu = muMesh StStatesY = roots(C(mu)); %solving W(y) = 0; StStatesY(imag(StStatesY) ~=0) = []; %deleting complex roots StStatesY(StStatesY < 0) = []; %deleting negative roots StStatesX = alpha*(1-beta*StStatesY); %calculating x coordinate indx = StStatesX < 0; StStatesX(indx) = []; %deleting steady states with negative x StStatesY(indx) = []; %deleting steady states with negative x for i = 1:length(StStatesY) %evaluating stability and plotting Jeval = J(StStatesX(i), StStatesY(i), mu); if all(real(eig(Jeval))<0) %point is stable plot(mu,StStatesY(i),'r.'); else plot(mu,StStatesY(i),'k.'); end end plot(mu, 0,'k.'); %always unstable semi-trivial steady state end hold off
After running the above code we should see a plot similar to the one below, but without the red dots that indicate the value of

We can see in the bifurcation diagram that for nominal
- if nominal, i.e. without treatment, value of
- if nominal, i.e. without treatment, value of
) then we can durably reduce the tumor to subclinical disease state, if during the treatment
This type of behavior is typically referred to as hysteresis. Most importantly this results is consistent with clinical observation that in some patients long lasting responses may be achieved even after the drug withdrawal.
From a mathematical point of view everything boils down to the question if we can force the trajectory to intersect the curve separating basins of attractions in the treatment free case. Once this is achieved we can withdraw the treatment and the patient’s immune system will take care of the disease by itself. Thus, for each initial condition in the phase space we should look for the value of 
First, we need to approximate the curve separating the basins of attraction. This can be achieved by the following MATLAB script:
%% DEFINING MODEL PARAMETERS sigma = 0.118; rho = 1.131; eta = 20.19; mu = 0.00311; delta = 0.374; alpha = 1.636; beta = 0.002; %% CALCULATING BIGGEST VALUE OF STEADY STATE FOR Y Smax = max(roots([mu*beta; -mu+beta*(eta*mu+delta-rho); ... sigma/alpha+rho-eta*mu-delta+beta*delta*eta; ... eta*(sigma/alpha-delta)])); %% DEFINING THE MODEL (INLINE FUNCTION) rhs = @(t,x)([sigma+rho*x(1,:).*x(2,:)./(eta+x(2,:))-mu*x(1,:).*x(2,:)-delta*x(1,:);... alpha*x(2,:).*(1-beta*x(2,:))-x(1,:).*x(2,:)]); %% FUNCTION RETURNING MODEL SOLUTION ON [0,100] FOR GIVEN INITIAL CONDITION solve = @(init)(ode45(rhs,[0 100],init)); %% CALCULATING LOWER PART OF THE SEPARATING CURVE yInit = 50; %initial value yTol = 1e-5; %tolerance dy = 5; %initial step size while dy > yTol sol = solve([0 yInit]); if abs(sol.y(2,end)-Smax) < 30 %reached maximal steady state yInitPrev = yInit; yInit = yInit-dy; else dy = dy/2; yInit = yInitPrev; end end %% CALCULATING UPPER PART OF THE SEPARATING CURVE xInit = 0.5; %initial value xTol = 1e-5; %tolerance dx = 0.5; %initial step size while dx > xTol sol = solve([xInit Smax+100]); if abs(sol.y(2,end)-Smax) < 30 xInitPrev = xInit; xInit = xInit+dx; else dx = dx/2; xInit = xInitPrev; end end %% CALCULATING THE FINAL CURVE sol2 = solve([0 yInit]); sol1 = solve([xInit Smax+100]); sol1.y = sol1.y(:,1:find(diff(sol1.y(2,:))>0,1,'first')); sol2.y = sol2.y(:,1:find(diff(sol2.y(1,:))<span data-mce-type="bookmark" id="mce_SELREST_start" data-mce-style="overflow:hidden;line-height:0" style="overflow:hidden;line-height:0" ></span><0,1,'first')); curve.x = [sol2.y(1,:) sol1.y(1,end:-1:1) ]; curve.y = [sol2.y(2,:) sol1.y(2,end:-1:1) ]; %% PLOTTING figure(1) clf plot(curve.x, curve.y)
Then having the curve we can implement the numerical solver in a way that it stops integration once the separating curve is reached. This is done by the following MALTAB script:
function sol = solveKuznetsovNondimensionalBasins( par, Tmax, init, curve )
init = init(:);
opts = odeset('RelTol',1e-8,'AbsTol',1e-8,'Events',@stops);
sol = ode45(@odesystem, [0 Tmax],init,opts);
function y = odesystem(~,x)
y = zeros(2,1);
y(1) = par.sigma+par.rho*x(1).*x(2)./(par.eta+x(2))-par.mu*x(1).*x(2)-par.delta*x(1);
y(2) = par.alpha*x(2).*(1-par.beta*x(2))-x(1).*x(2);
end
function [value,isterminal,direction] = stops(~,y)
value = y(2)-interp1(curve.x, curve.y, y(1));
isterminal = 1; % Stop the integration
direction = 0; % Any direction
end
end
Using the above procedures we can look at how trajectories starting from the same initial condition (initial point) will behave for different
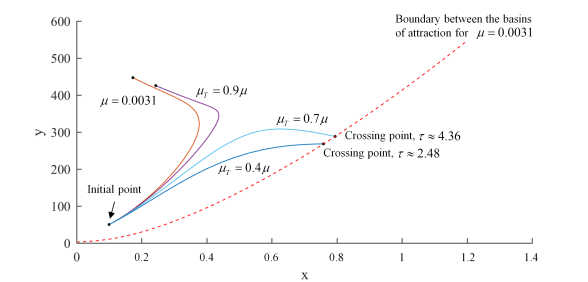
In the above plot we see that if the drug dose is not sufficient (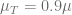
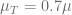
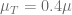
Finally, for each initial point we can evaluate if for a given dose (value of

As it can be seen in the above plot, the possibility to observe therapeutical success depends strongly on the dose of the drug, time on therapy, and initial T cell infiltration. It is conceivable that the above simple model, if successfully calibrated with patient data, could augment patient-specific treatment design.
 and
and  ) with different growth rates (
) with different growth rates ( ) that compete for the limited amount of space (K) and respond differently to treatment (
) that compete for the limited amount of space (K) and respond differently to treatment ( ):
):
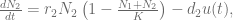
 describe drug concentration and under usual pharmacokinetic assumptions is expressed as
describe drug concentration and under usual pharmacokinetic assumptions is expressed as
 is the drug dose,
is the drug dose,  is drug administration moment, and
is drug administration moment, and  is clearance rate of the drug.
is clearance rate of the drug. ) at simulation endpoint:
) at simulation endpoint: times for randomly generated parameters in the for loop:
times for randomly generated parameters in the for loop:



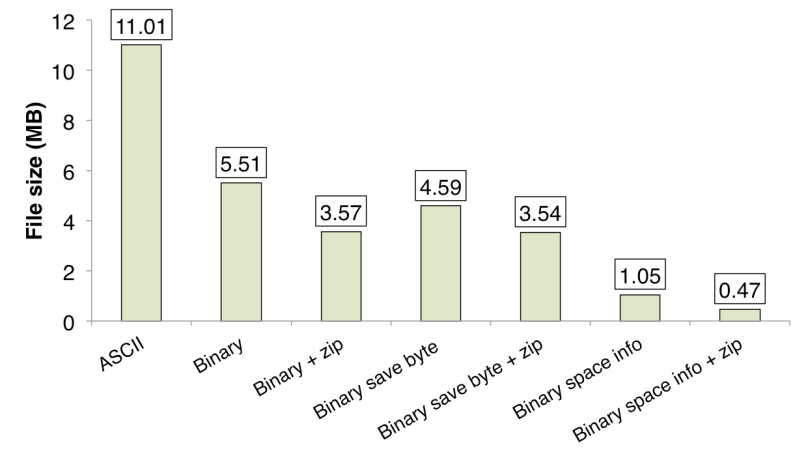 Figure 1. Comparison of the amount of used disk space.
Figure 1. Comparison of the amount of used disk space.





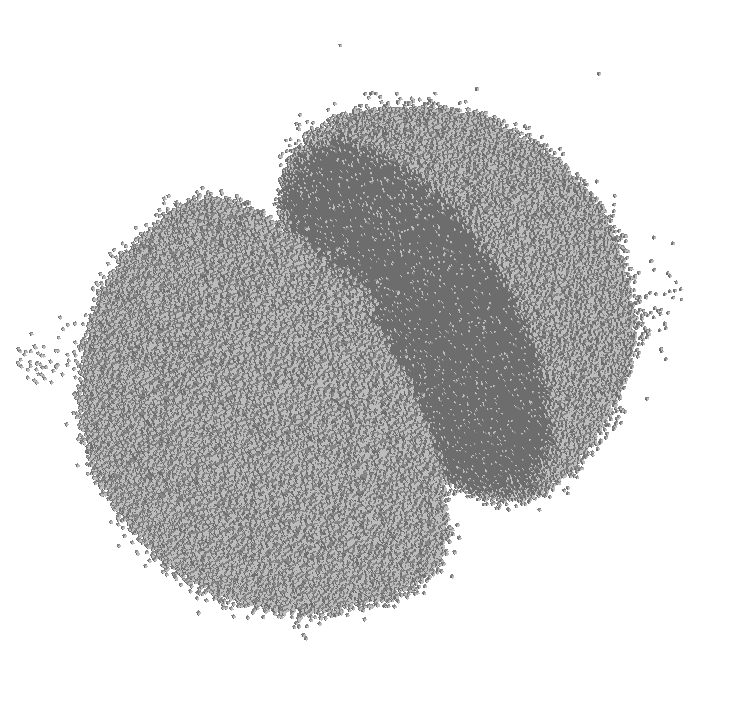














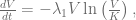
 represents spontaneous loss of functional vasculature,
represents spontaneous loss of functional vasculature,  represents vessels growth stimulation due to factors secreted by the tumor proportionally to its size, and
represents vessels growth stimulation due to factors secreted by the tumor proportionally to its size, and  describes endogenous inhibition of previously generated vasculature due to factors secreted by the tumor proportionally to the tumor surface-to-volume ratio.
describes endogenous inhibition of previously generated vasculature due to factors secreted by the tumor proportionally to the tumor surface-to-volume ratio.


 . It seems that there is nothing more to do – we estimated the parameters and nicely fitted model to the experimental data. However…
. It seems that there is nothing more to do – we estimated the parameters and nicely fitted model to the experimental data. However…







{kind=link}